ZincBindPredict—Prediction of Zinc Binding Sites in Proteins
This paper is the final publication to come out of my PhD research, and addresses the core question that the project has focused on.
It's about zinc binding sites. Proteins - the big, complex molecules that do almost everything of note in biology - sometimes have metal atoms stuck to them which enable them to do their job. They first fold up without that metal atom, but as a consequence of the way they fold up, they end up with a region of their surface that a particular metal atom will be attracted to, because of the region's shape or the atoms involved. That region of the protein that the metal atom is attracted to is the 'metal binding site' - and when it's zinc that is attracted, it is a zinc binding site. Lots of proteins have zinc binding sites, and therefore have a zinc atom associated with them as a consequence.
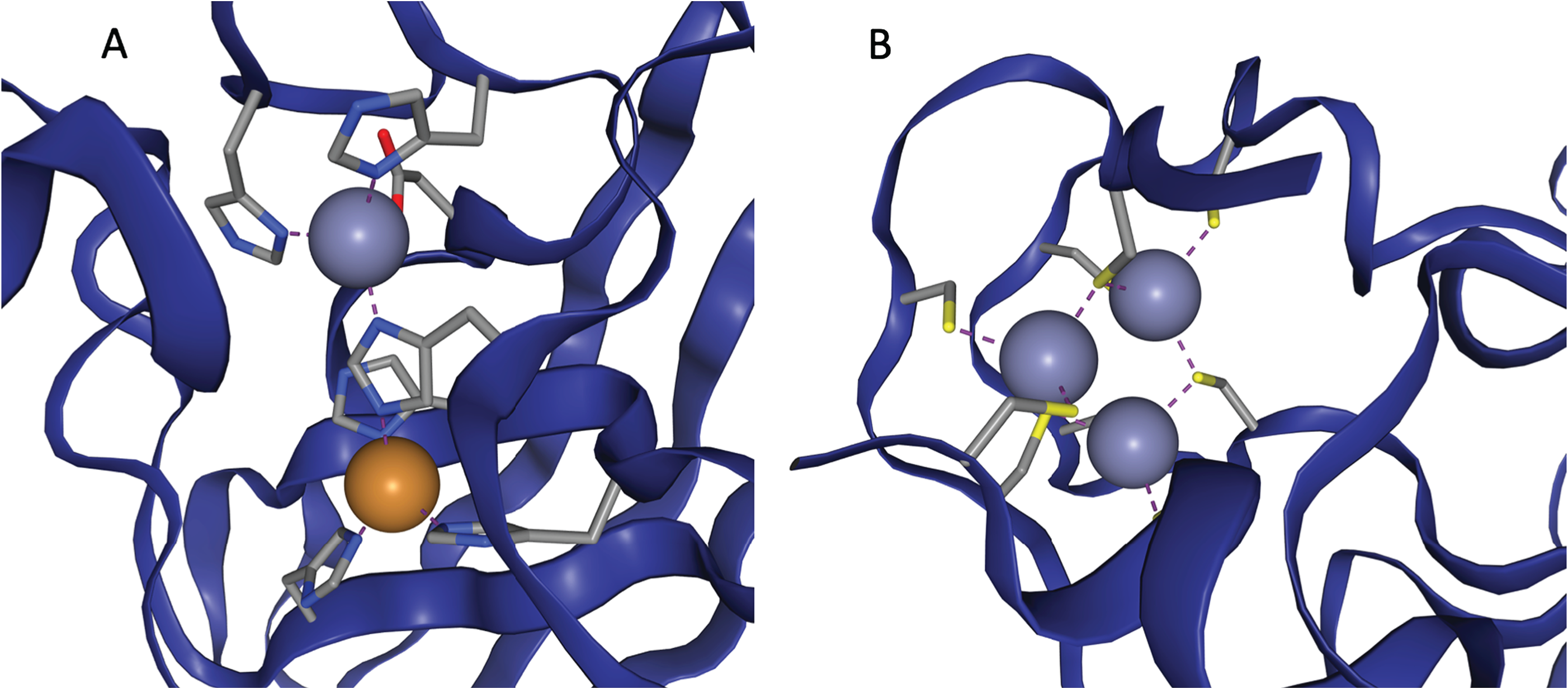
Two examples of zinc binding sites, taken from ZincBind.
Earlier in the PhD, I created ZincBind - a database of all the zinc binding sites we currently know about, as well as their properties. The paper on this was published two years ago, and the resource has been automatically updated every week ever since then, to the point where it currently has nearly 40,000 zinc binding sites in it - all of them annotated with information about the binding site itself. This resource is publicly available, has some helpful visualisation tools, and is (hopefully) useful to research in zinc biology generally.
For my purposes however, there was a very specific reason for creating that database. The second half of my PhD has focused on predicting zinc binding sites. That is, you take a protein (either its 3D structure before zinc is bound, or just the sequence of its 'residue' subunits) and, based on what we know about zinc binding sites, predict whether any part of that protein could bind a zinc atom. Being able to do this well brings a lot of benefits - there are still many proteins out there whose function is unknown, and if you can at least say if it would bind zinc, you can begin to infer what it does. And even for proteins whose function is well understood, understanding where and how it binds zinc (especially if it does so in disease states) can offer insight in how the protein works, and how to design drugs to target it.
So - the intention was to use the big dataset of known zinc binding sites I had previously built to determine what the typical properties of zinc binding sites are, and use those properties to hunt for zinc binding sites in other proteins. One way of doing this might be to do so manually - you would literally make a list of observed properties, such as 'atoms tend to approach at an average of X nm except in circumstance Y...' etc., and then write a computer program with all of these rules encoded in it - which would then look for these properties in other proteins.
However in practice that would be insufferably tedious and probably not all that useful, as it relies on a human (me) being able to spot all the properties and express them statistically. Some of the properties may be very subtle, and in any case there may be too many to manually identify. Instead machine learning is used. Despite the voodoo and woowoo around that term, here it simply means automating the process of working out what the properties of zinc binding sites are. I created a list of zinc binding sites (with key properties picked out such as particular atom distances), a list of residue combinations that weren't zinc binding sites, and fed them to a machine learning algorithm which identifies what properties can distinguish zinc binding sites from non-zinc binding sites. There is a lot more technical detail to this which the paper goes into, but for here it is sufficient to say that the particular algorithm I used - Random Forest - works by creating a sort of decision tree, where it learns what cutoffs to look for in the different measurements to allocate a given residue combination into the 'zinc binding' bin or 'not zinc binding bin'. No human frailty here - just mathematics applied to a large dataset.
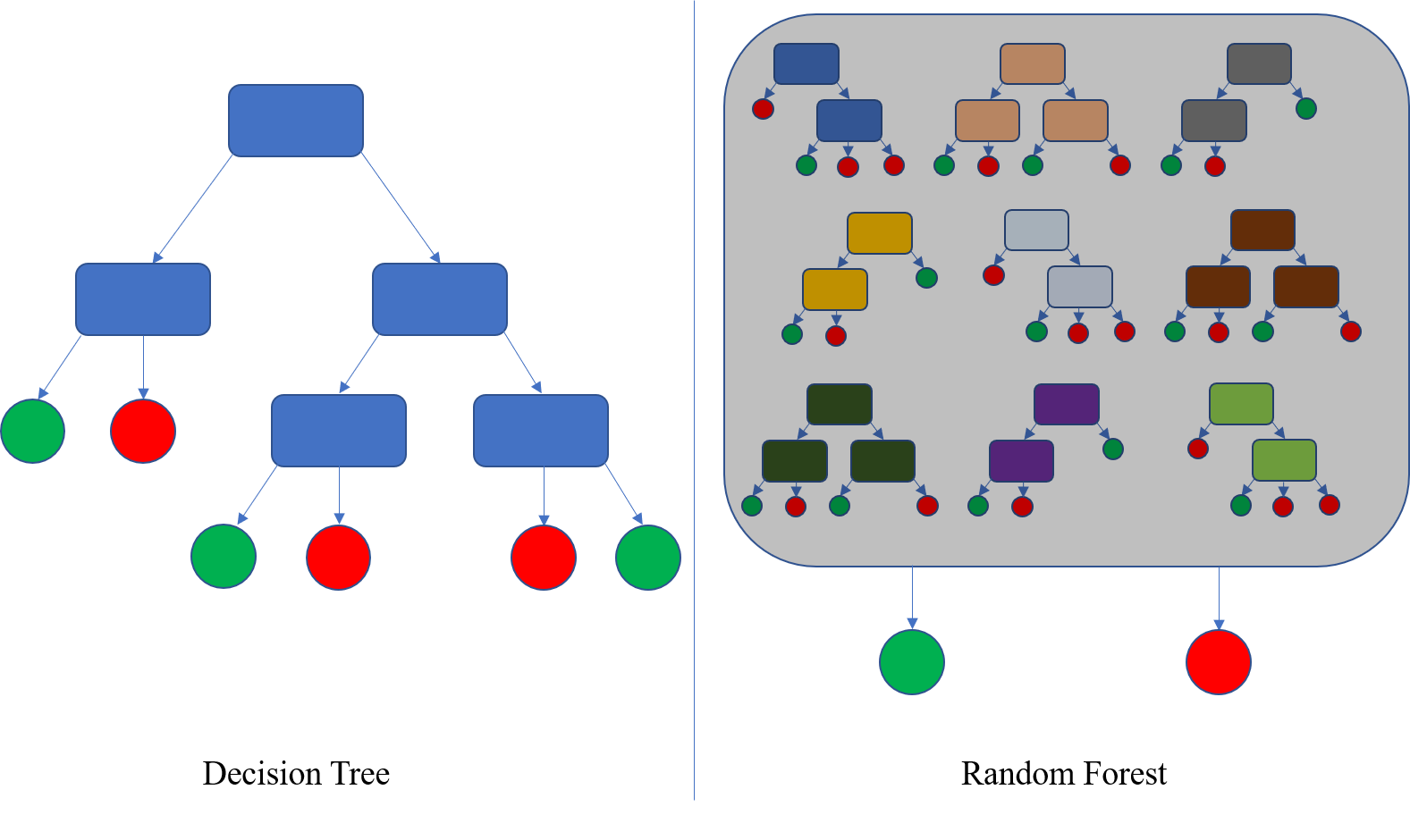
An overview of how Random Forest works, from wikipedia. On the left is a single decision tree, that allocates inputs into one of two categories (red and green here - sorry colour-blind people). A random forest uses multiple decision trees.
All this has been tried before of course, to varying degrees of success - I am not the first to create a tool which predicts zinc binding. One crucial difference with my approach however, is that there isn't simply one zinc binding site predictor - instead I divided them into different distinct subtypes (based on the kinds of residue involved) and created highly specific predictors for each of those subtypes. This made them perform extremely well - in the high 90s for most meaningful metrics. The paper goes into more technical detail about how these are validated, but the salient point is this - if you train predictive models on specific sub-types of zinc binding site and use them in aggregate to predict zinc binding sites, you get vastly superior results than if you just look at zinc binding in generally. Of course there's a slight tradeoff, in that only the most common sub-types can be predicted now because the less frequent sub-types don't have enough data to train a predictive model - but that's a temporary problem as the database is always growing.
The final tool - ZincBindPredict - is available here or alternatively as a GraphQL API.
Ultimately, this result makes it easier to predict zinc binding in proteins, and consequently improves our ability to annotate the whole biological universe of proteins and understand how it all works. In a world where no zinc binding site goes undiscovered, our understanding of how proteins work is improved, and so is our ability to intervene when they go wrong.