Thermal melt circular dichroism spectroscopic studies for identifying stabilising amphipathic molecules for the voltage-gated sodium channel NavMs
This was my first ever scientific paper, and was the result of a rotation I did during the first year of my PhD in Professor Bonnie Wallace's lab.
Biologists spend a lot of time and energy (and money) studying proteins. Proteins are arguably the single most important class of molecule in living things - pretty much every function is carried out by some kind of protein or other. So being able to work out the actual atomic structure and layout of a particular protein, or what it does, or even just being able to isolate it from the rest of the chemicals inside a living thing - is hugely important. Medical advances rely on this.
But it's also very difficult. All proteins require many hours or days of work in the lab to even get them into a state and an environment where they can be studied. And some proteins require more work than others.
Membrane proteins are a particular class of protein, which live in the membranes of cells - the fatty outer layer of cells that separates them from the outer environment. They are a very important class of protein - many of the proteins that our drugs act on are membrane proteins (perhaps unsurprising, since they are accessible from outside the cell) - so being able to study them is vital. Unfortunately they are very difficult to work with, even by the already exhausting standards of regular protein purification.
Why? Well there aren't very many of them in a cell for one. When you can only live in the thin outer edge of a cell, there will naturally be fewer of you than a protein that can go anywhere in the cell. So this means that to get enough protein to study, you have to grow a lot of cells - litres usually.
Secondly, proteins can't be studied while still in the membrane - it just gets in the way of the various techniques that Biologists use to study them. So extracting them from the membrane has to be done. All proteins have to be extracted from the cell they were grown in, but with membrane proteins it's a particular hassle because you also have to also remove them from the membrane they are stuck in.
Finally, whereas with most proteins you can just remove them from the cell and put them in (more or less) water - this won't work with membrane proteins. The useful things that a protein does relies on it being folded up in a particular way, and proteins just fold differently when they are in a membrane than when they are in solution. So when they are removed from the membrane and put into water, their fold changes, and so they will no longer 'work'. Because of this, membrane proteins are not just taken from the membrane and put into a watery solution - they are first wrapped in something else which resembles the membrane they have just been ripped from. This is called solubilising, because it enables the protein to float freely through water with a protective outer layer to maintain it in the proper fold.
Often, detergents are used for this last step. Like membranes they are fatty, oily substances, which form very small spherical balls in solution called micelles. Proteins which normally live in cell membranes can also live in these micelles because they are fairly similar to membranes. The vast majority of scientific studies that have been done on membrane proteins - be they experiments to work out the 3D structure of the proteins, or experiments to measure what they do physiologically - have been done using detergent molecules.
Detergents are not perfect however. For one thing, while they are pretty similar to membranes, they are not exactly the same, and for this reason there will be small changes in the protein's fold when studied this way. It will be close enough that you can still work out the 3D structure of the protein, but not close enough for the protein to still do its job.
Another problem is the slightly more complex issue of excess detergent. Not all detergent molecules in a solution will form micelles - there will always be some fraction that just float around as individual molecules. In fact if the concentration of detergent falls below some specific concentration, the micelles will just fall apart. For this reason, you always need to have 'extra' detergent molecules in your solution in order to keep the micelles intact, and these otherwise-useless molecules can interfere with any experiment you might be doing.
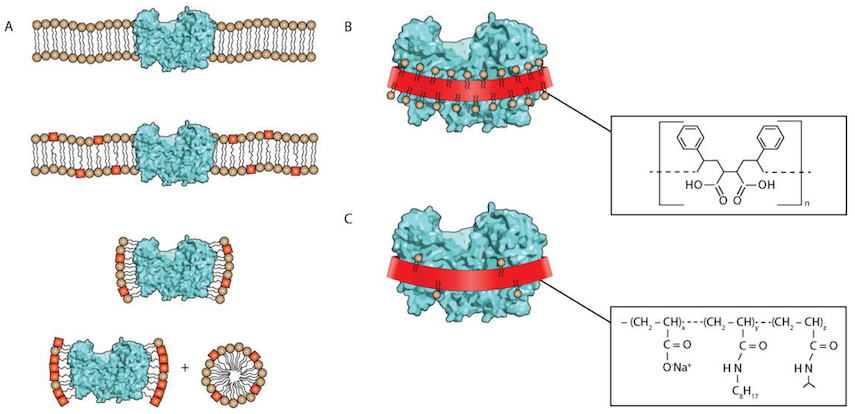
So, scientists are always on the lookout for alternatives to detergents. One alternative that was invented in the 1990s, are amphipols.
Amphipols are 'polymers' - that means that a molecule of an amphipol is a very long chain made up of many repeating units, each repeating unit being about the same size as an individual detergent molecule. They also have 'side chains' - bits that stick out from the backbone of the chain. These side chains are fatty and oily like detergents and membranes, whereas the backbone is not. So they can wrap around proteins, and the side chains provide a nice membrane-like environment for the protein, while the backbone allows the whole thing to dissolve in water.
Taken from this review.
Amphipols seem to have some advantages over detergents. For reasons that are not entirely clear, they do a lot better job at maintaining the protein in the correct fold - to the point where proteins will usually still be able to carry out their function in amphipols whereas they often don't in detergents. Amphipol chains will also stick to proteins regardless of their concentration, which means any excess amphipol can be removed before they are studied - again this can't be done with detergents.
However, for amphipols to be considered a viable alternative to detergents, they need to be much more rigorously tested. After all, detergents may have their faults, but they have been widely used for a long time, and we know their behaviour under all kinds of conditions.
One area where amphipols had not yet been tested against detergents is their ability to maintain the fold and structure of membrane proteins at higher and higher temperatures. When any molecule - including proteins - get hot, their atoms jiggle around a lot more vigorously. The forces between the atoms that keep the protein in its proper fold are overcome by this jiggling as the temperature increases, and at a certain temperature (it will vary from protein to protein) the whole thing will just unravel.
The detergent or amphipol wrapped around a membrane protein can stabilise the protein and prevent this from happening - although even in the most ideal situation there will always be some temperature at which the protein cannot hold together. But some solubilising agents might be better than others.
So, that is what we looked at in this study. We compared two detergents and two amphipols on their ability to stabilise the structure of a membrane protein at higher and higher temperatures.
What We Did
The membrane protein we used is called 'NavMS'. It comes from a species of bacteria called Marinococcus marinus, but the protein is closely related to a protein in humans which is a common drug target, and which serves an important role in neurone firing. The full 3D structure of this protein had already been solved, so we knew what we were starting with.
The two detergents we used are called 'DDM' and 'Cymal-5' - both fairly common detergents in membrane protein science, and both fairly similar to each other in structure. The two amphipols we used were 'A835' and 'Pmal-C8' - again these are fairly commonly used amphipols.
The way we measured how unfolded the protein was at a given temperature was by measuring its 'alpha helicity'. Alpha helices are coils in the strand that makes up proteins, and the more unfolded the protein gets as the temperature rises, the more these will unravel and become disordered. You can measure how much alpha helicity a protein has by using a technique called circular dichroism spectroscopy. This shines a specific kind of light through a sample of protein, and by examining the specific wavelengths of light that get absorbed after it has passed through the sample, you can calculate the alpha helicity of the protein in that sample.
We used a special kind of circular dichroism experiment called a thermal melt - this is just like a normal circular dichroism experiment except that after it is complete, it raises the temperature by a fixed amount and scans the sample again. This repeats until the maximum temperature is reached. In our case we started at 20 °C and increased this to 80 °C in 5 °C increments.
So, to summarise - amphipols might be a useful alternative to detergents as 'solubilising agents', molecules that allow scientists to study membrane proteins, and so the more we know about these amphipols, the more useful they will be in pharmacology, structural biology, and many other areas of research. We decided to investigate one particular property of them - how well they stabilise proteins at high temperatures, compared with detergents. We did this by using thermal melt circular dichroism to examine how each molecule maintained the alpha helicity of a particular membrane protein.
The exact details of how we grew a big vat of cells producing the NavMS protein, extracted the protein from them, and solubilised them in one of the four solubilising agents being tested, are in the actual paper. We purified them all to a concentration of about 1 mg/ml, as that is the optimal concentration for the circular dichroism machine we used.
The final step in preparation is a concentration step, where you get the sample up to that concentration by taking what is usually a much more dilute sample and passing it through a concentrator - essentially a tube with a filter, which you spin very fast to force liquid through the filter - the protein is too big to pass through, so it remains in the top compartment in an ever shrining amount of liquid. The liquid that passes through is kept as flowthrough - this is useful because it is identical to the sample except that it doesn't have the protein in it - which is important because when you scan a sample with circular dichroism, you can't know how much of the effect you see is because of your protein, and how much is because of the other stuff floating around in there (things like salts, and other chemicals used in the preparation of the sample). To solve this problem, you need a baseline to scan too, so that you can subtract it from the output of the 'real' experiment. The flowthrough is perfect for this because it should contain everything that the sample contains, except for the protein itself (and its surrounding solubilising agent obviously).
A single circular dichroism experiment takes a whole day in this case. A single scan will only take about ten minutes, but a thermal melt can take much longer - it has to scan the sample, wait for the temperature to be raised by 5 °C, scan again, raise the temperature again… The whole thing can take about an hour. Add to that the fact the baseline and sample need to be scanned on the same day, and it quickly ends up taking up the whole day. We did multiple thermal melt experiments on each solubilising agent, over about a month and a half.
The Results
We processed the data using a program called CDtool and used the processed data to estimate alpha helicity using a program called DichroWeb. Both of these programs were created and are maintained by the lab I did this rotation in.
The output of a thermal melt looks like this:

Here the top-left represents Pmal-C8, the top-right is A835, bottom-left is DDM and bottom-right is Cymal-5. The black line in each represents the scan that was done at 20 °C. The way circular dichroism works is that you shine left-handed and right-handed circularly polarised light through the sample at different wavelengths of light, and look to see if there is a difference in absorbance between left and right handed light at each wavelength. So in these cases, there is no difference at 280 nm, at 270 nm, at 260 nm etc. At 230 nm one kind is absorbed more than the other, and by 195 nm the other kind of light is absorbed more.
(To be honest I forget whether negative values mean left handed predominates over right, or vice versa, but you get the idea.)
The lines then go from green to red as the temperature of the scan goes from 25 °C to 80 °C (the dotted black line represents a cool down back to 20 °C). If the protein was completely unaffected by the rising temperature, you would expect no change in the output lines in these charts, because a change in alpha helicity would lead to a change in absorption, and so a change in the lines in the chart. In all cases, as expected, rising temperature does affect alpha helicity.
Just looking at them by eye, a difference is apparent. The lines in the Pmal-C8 chart are very close together, suggesting not much changes for the protein when it has the amphipol Pmal-C8 wrapped around it. On the other hand, the lines are very far apart in the case of Cymal-5, suggesting that it is doing a particularly awful job of holding the protein together under rising temperatures. A835 and DDM look pretty similar.
To really assess what is happening though, you need to numerically determine the extent of alpha helix loss at rising temperatures. This is what the program DichroWeb does. You feed it the data represented in the chart above, and it gives you the following data:

This time, each line represents one of the four agents - Pmal C8 (red), A835 (blue), DDM (green) and Cymal-5 (purple) - and the change in alpha helicity as temperature rises is shown more clearly.
The story it tells is pretty much the same. The amphipol Pmal C8 manages to keep the protein stable and pretty much unchanged until the very highest temperatures, the amphipol A835 and the detergent DDM are about the same, and not quite as good as Pmal C8. They are both better than the detergent Cymal-5 however, which lets the protein unravel as soon as the temperature gets to 50 °C.
It's also worth noting that in each case the protein starts out with the same alpha helicity, just under 70%, which is closely in agreement with the previously estimated alpha helicity for this protein. This all suggests that all four solubilising agents do am equally good job of maintain the natural fold of the protein at the start, even if they have varying abilities to maintain it as temperature rises.
So, clearly there is a difference in usefulness between the four compounds - at least in terms of their ability to maintain the structure of the membrane protein at high temperatures. It is not a straightforward case of amphipols always being better than detergents however. The best candidate was an amphipol, and the worst was a detergent, but in the middle you have the amphipol A835 and the detergent DDM which are practically indistinguishable.
Certainly Pmal-C8 shows promise as a potential replacement for detergents for some purposes, but some amphipols don't perform particularly better than common detergents.
This was my first academic publication, and I am hugely grateful to Prof. Wallace and her lab - especially Altin, who helped me every step of the way and who is my co-author on this paper.